
Definição
Os distúrbios da oxidação dos ácidos graxos (DOAG) são deficiências genéticas metabólicas nas quais o organismo é incapaz de oxidar os ácidos graxos para produzir energia, devido à ausência ou mau funcionamento de uma enzima específica.
A principal fonte de energia para o organismo é a glicose, no entanto quando a glicose se esgota, a gordura é oxidada para produzir energia. Crianças e adultos com “DOAG” não possuem esta disponibilidade de energia prontamente.
A via metabólica das gorduras é a principal fonte de energia para os músculos esqueléticos e cardíaco, enquanto o fígado a utiliza principalmente durante períodos de jejum, quando os ácidos graxos se tornam o principal substrato para a produção de energia neste órgão. O cérebro, embora não oxide os ácidos graxos, utiliza os corpos cetônicos, derivados da acetil-CoA e da acetoacetil-CoA produzidas pela beta-oxidação hepática dos ácidos graxos.
Os principais fenótipos clínicos dos “DOAG” são: a hipoglicemia não cetótica (hipocetótica), a cardiomiopatia e a miopatia. Embora estas alterações possam estar presentes simultaneamente em alguns destes distúrbios, um dos fenótipos em geral é o predominante.
O perfil de acilcarnitinas é fundamental na investigação dos transtornos da beta-oxidação mitocondrial dos ácidos graxos.
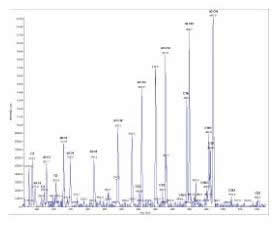
Fig. 2 Perfil de acilcarnitinas em amostra de sangue em papel-filtro de recém-nascido normal. Espectrômetro de massas em tandem em modo de operação “precursor ion scan”. Após volatilização/ ionização da amostra, os íons precursores são filtrados pelo primeiro quadrupolo Q1, ajustado para a faixa de massas 250-550 u.m.a. (unidade de massa atômica), e entram na câmara de colisão, onde são fragmentados. Q3 permanece fixo em 85 u.m.a. O espectro corresponde às massas de todos os pares transicionais que obedecem à seguinte equação: íon produto = 85 u.m.a. e íon precursor = 250-550 u.m.a. Os picos dos padrões internos, que são acilcarnitinas marcadas com deutério, estão identificados por d (o número após d indica o número de átomos de deutério). O número após C indica o número de átomos de carbono, e as insaturações são apontadas pelo algarismo em seguida aos dois pontos, assim C18 é a octadecanoilcarnitina (estearoilcarnitina) e a C18:1 é a octadecenoilcarnitina (oleilcarnitina).
Para interpretarmos a fisiopatologia dos distúrbios da beta-oxidação dos ácidos graxos, é essencial entender esta via metabólica. Para sofrerem a beta-oxidação, os ácidos graxos são ativados pela ação das acil-CoA-sintetases, formando tioésteres de CoA (acil-CoA) 8. Embora os ácidos graxos de cadeia curta ou média atravessem livremente as membranas mitocondriais, o mesmo não se dá com os ácidos graxos de cadeia > 10 carbonos, que requerem o ciclo das carnitinas para o seu transporte até a matriz mitocondrial (fig 3). Foram descritos transtornos por deficiência das enzimas do ciclo da carnitina: transportador de carnitina (CT), carnitina- palmitoiltransferase I (CPT 1), translocase e carnitina-palmitoiltransferase II (CPT 2).
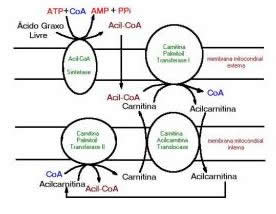
Fig. 3 Transporte dos ácidos graxos de cadeia longa do citoplasma até a matriz mitocondrial onde são oxidados. Após a ativação dos ácidos graxos livres em ésteres de CoA (acil-CoA), há uma troca da CoA pela carnitina por ação da carnitina-palmitoiltransferase I (CPT I). As acilcarnitinas são então transportadas para o interior da mitocôndria, onde ocorre uma troca reversa através da ação da carnitina-palmitoiltransferase II (CPT II). Uma vez no interior da mitocôndria, as acil-CoA serão o substrato da beta-oxidação (figura de Michael W. King, Ph.D., Indiana State University School of Medicine).
A via metabólica é denominada beta-oxidação porque se processa pela remoção seqüencial de uma unidade de 2 átomos de carbono (molécula de acetil-CoA) pela oxidação do carbono na posição beta da molécula de acil-CoA. A primeira etapa envolve a oxidação da acil-CoA em enoil-CoA, catalisada pelas acil-CoA desidrogenases FAD-dependentes. Quatro acil-CoA desidrogenases FAD-dependentes foram descritas em seres humanos: VLCAD, LCAD, MCAD e SCAD. Estas enzimas são definidas pelo número de carbonos das acil-CoA sobre as quais atuam: VLCAD (“very long-chain acyl-CoA dehydrogenase”: acil-CoA desidrogenase de cadeia muito longa): C20 a C14; LCAD (“long-chain acyl-CoA dehydrogenase”: acil-CoA desidrogenase de cadeia longa): C18 a C12, ótimo: C16; MCAD (“medium-chain acyl-CoA dehydrogenase”: acil-CoA desidrogenase de cadeia média): C12 a C4, ótimo: C8; SCAD (“short-chain acyl-CoA dehydrogenase”: acil-CoA desidrogenase de cadeia curta): C6 e C4.
Deficiências de todas estas quatro enzimas (VLCAD, LCAD, MCAD e SCAD) já foram descritas em seres humanos. A deficiência de MCAD é o mais bem estudado dos transtornos da beta-oxidação dos ácidos graxos, tendo ganhado alguma notoriedade pela sua associação com a síndrome da morte súbita do lactente. É uma doença autossômica recessiva que apresenta maior incidência em brancos de origem norte-européia (Reino Unido, Holanda, Países Escandinavos, Alemanha).
Uma mutação pontual, 985A Ü G (adenina substituída pela guanina na posição 985) no éxon 11 do gene da MCAD representa 90% dos alelos causadores da deficiência de MCAD. A incidência varia de 1:8.930 a 1:18.500 nascidos vivos em estudos de triagem neonatal, realizados no Reino Unido e EUA.
Tipicamente, a deficiência de MCAD se manifesta na forma de episódios agudos intermitentes de hipoglicemia hipocetótica (não necessariamente não-cetótica), aumento do “anion gap”, hiperuricemia, e discreta hiperamonemia, que se expressam por vômitos, letargia, e alterações mentais incluindo o coma. Pode haver elevação das transaminases e outros sinais de insuficiência hepática. Este quadro clínico pode levar à confusão diagnóstica com a síndrome de Reye, podendo culminar com coma e morte.
Durante estes episódios são excretadas pela urina grandes quantidades de ácidos ômega-dicarboxílicos de cadeia retilínea com número médio de átomos de carbono, C6 a C10 (dicarboxilicacidúria) 12, que podem ser demonstrados por cromatografia gasosa. Estes ácidos são formados pela ômega-oxidação dos ácidos monocarboxílicos de 10 a 12 átomos de carbono, que são posteriormente encurtados pela beta-oxidação a C6-C10.
O perfil de acilcarnitinas plasmático obtido por MS/MS mostra caracteristicamente um aumento de octanoilcarnitina (C8).
Existe um outro subgrupo dos transtornos da beta-oxidação dos ácidos graxos formado pelas deficiências de 3-hidroxiacil-CoA desidrogenases NAD+-dependentes: SCHAD (“short-chain 3-hydroxyacyl-CoA dehydrogenase”: 3-hidroxiacil-CoA desidrogenase de cadeia curta), que atua sobre uma ampla faixa de compostos 3-hidroxi-acil-CoA, que vão de 16 a 4 átomos de carbono [a designação “short chain” (cadeia curta) é de certa forma inadequada] e LCHAD (“long-chain 3-hydroxyacyl-CoA dehydrogenase”: 3-hidroxiacil-CoA desidrogenase de cadeia longa), associada à proteína trifuncional (LCHAD + 2-enoil-Co A hidratase + beta-cetotiolase).
Sintomas
É muito ampla a forma de apresentação dos “DOAG”. Na tabela abaixo podem ser encontrados os fenótipos clínicos associados aos diversos “DOAG”.
|
Fenótipos Observados nos Distúrbios da Oxidação dos Ácidos Graxos |
|
| Distúrbios do Ciclo da Carnitina | |
| Deficiência | Fenótipo(s) |
|
Transportador de Carnitina (CT) |
• Cardiomiopatia dilatada |
|
Carnitina palmitoil transferase I |
• Hipoglicemia hipocetótica |
|
Translocase |
• Cardiomiopatia hipertrófica com hipoglicemia hipocetótica |
|
Carnitina palmitoil transferase II |
• Cardiomiopatia neonatal com hipoglicemia hipocetótica e morte na 1a semana de vida |
| Distúrbios da Membrana Mitocondrial Interna | |
| Deficiência | Fenótipo(s) |
|
VLCAD |
• Hipoglicemia hipocetótica |
|
LCHAD |
• Hipoglicemia hipocetótica com miopatia esquelética |
|
Proteína Trifuncional |
• Cardiomiopatia, miopatia e hipoglicemia hipocetótica |
| Distúrbios da Matriz Mitocondrial | |
| Deficiência | Fenótipo(s) |
|
MCAD |
• Hipoglicemia hipocetótica |
|
SCAD |
• Atraso do desenvolvimento, hipotonia, (convulsões, microcefalia) |
As crises de hipoglicemia hipocetótica são situações de emergência que sobrevêm em resposta tanto ao jejum prolongado (por exemplo, o desmame da mamada noturna) quanto a infecções comuns intercorrentes (por exemplo, viroses gastrointestinais ou infecções de vias aéreas superiores), que tipicamente causam perda de apetite e aumentam o consumo de energia, devido à febre. Outros sintomas podem incluir vômitos, diarréia, letargia, convulsões, coma e dificuldade respiratória.
Hepatomegalia e doença hepática aguda estão quase sempre presentes durante uma crise de hipoglicemia hipocetótica, que também é caracterizada por aumento do “anion gap”, hiperuricemia, elevação das transaminases hepáticas, e discreta hiperamonemia. Este quadro clínico pode levar à morte súbita e inexplicável em um número considerável de pacientes, levando erroneamente ao diagnóstico de Síndrome da Morte Súbita do Lactente (SMSL) ou de Síndrome de Reye. Uma deterioração clínica rápida, desproporcional para uma simples e benigna infecção, pode levar à suspeita de “DOAG” e requer a imediata administração de glicose intravenosa e coleta de amostras de sangue e urina para testes metabólicos.
Diagnóstico e tratamento
Os “DOAG” são doenças autossômicas recessivas que afetam ambos os sexos. Os pais de uma criança afetada são obrigatoriamente heterozigotos para o alelo anormal existindo uma probabilidade de 25% de cada novo filho do casal ser afetado por um “DOAG”. Se uma criança for diagnosticada como apresentando um “DOAG”, seus irmãos deverão ser testados mesmo se forem assintomáticos.
O diagnóstico laboratorial quase sempre inclui perfil de acilcarnitinas quantitativo do plasma, análise de ácidos orgânicos urinários, dosagens de carnitina total e livre e ensaios enzimáticos em fibroblastos. O perfil de acilcarnitinas quantitativo do plasma ou sangue total em papel-filtro é a forma mais direta de diagnóstico da maioria dos “DOAG”. Análise de DNA para pesquisa de mutações também é útil, porém está disponível somente para as mutações mais comuns das deficiências de MCAD e LCHAD, por enquanto.
Recomenda-se tanto no caso de deficiência de MCAD quanto de LCHAD, fazer a análise simultânea do perfil de acilcarnitinas quantitativo e a análise do DNA para pesquisa da mutação em amostra de sangue total em papel-filtro.
O tratamento para “DOAG” envolve diversas abordagens. O mais importante é evitar o jejum por 4 – 6 horas. Um período de jejum, especialmente quando associado a uma enfermidade infecciosa pode desencadear uma “crise metabólica” levando à hipoglicemia e letargia, necessitando de hospitalização. Se a criança for hospitalizada é imperativo, de acordo com especialistas em “DOAG”, que seja iniciado de imediato glicose a 10% endovenosa, logo após a coleta de sangue para análises bioquímicas.
Múltiplas refeições com alimentos pobres em gordura e ricos em carboidratos (como cereais, massas e bebidas açucaradas) são recomendadas ao longo do dia. Crianças abaixo de um ano deverão ter pelo menos uma alimentação à noite para evitar que fiquem 12 horas em jejum. Para ajudar a reduzir a frequência de hipoglicemia pela manhã, sugere-se misturar 01 a 03 colheres de sopa de amido de milho (maisena) a uma bebida fria ou alimento frio servido à noite. É importante que a maisena não seja cozida.
Suplementos de L-carnitina são essenciais na deficiência do transportador de carnitina (CT), mas podem ser utilizados nos outros casos de “DOAG”, para corrigir a deficiência secundária de carnitina frequentemente observada e aumentar a eliminação de metabólitos tóxicos. Embora não tenham sido provados os benefícios concretos destes suplementos na maioria dos “DOAG”, a dose recomendada de carnitina oral é de 100 mg/kg/dia.
Na deficiência de MCAD não foi observado aumento da destoxificação de ácidos graxos de cadeia média mediada pelos suplementos de carnitina, avaliada pela excreção urinária de acilcarnitinas de cadeia média. Por outro lado, não foram relatados efeitos colaterais nocivos dos suplementos de carnitina em pacientes com deficiência de MCAD, em contraste com a deficiência de LCHAD, onde a formação de variedades de 3 hidroxiacilcarnitinas de cadeia longa é tida por alguns autores como prejudicial.
Aspectos Clínicos de Alguns Distúrbios da Beta-Oxidação dos Ácidos Graxos
Neste grupo as doenças se manifestam pela incapacidade metabólica de oxidar ácidos graxos por ausência ou disfunção enzimática, e em outros casos por esgotamento secundário de carnitina.
- Deficiência de carnitina palmitoil transferase tipo II (CPT II)
- A CPT II afeta mais o sexo masculino que o feminino e é mais aparente nas pessoas com diabetes ou deficiência nutricional. O jejum pode desencadear os sintomas, cujos mais importantes são mialgia, fadiga e urina marrom-avermelhada.
- Normalmente torna-se aparente em adultos, mas uma forma mais severa afeta crianças.
- O tratamento inclui uma dieta restrita em proteínas e gorduras e rica em carboidratos, hidratação adequada, evitar jejum e manter a criança aquecida.
- A suplementação de carnitina pode ser eficaz.
- Deficiência de múltiplas acil-CoA desidrogenase
- A Deficiência de múltiplas acil-CoA desidrogenase também conhecida como Acidemia Glutárica Tipo II (GA II) manifesta-se sob três formas, sendo a forma neonatal bastante severa e frequentemente fatal dentro de algumas semanas.
- Os sintomas de GA II neonatal em crianças com anomalias congênitas podem incluir hipoglicemia severa, acidose metabólica, hipotonia, hepatomegalia e frequentemente um odor de “pés suados”.
- Quando as anomalias congênitas estão ausentes, os sintomas podem ser mais leves e crianças não tratadas podem sobreviver durante um período mais longo.
- O tratamento inclui uma dieta rica em carboidratos, restrita de proteínas e gorduras, com refeições frequentes. A suplementação com riboflavina e carnitina pode ser útil.
- Deficiência de desidrogenase de 3-hidroxi-acil-CoA de cadeia longa (LCHAD)
- Sintomas típicos de LCHAD são hipoglicemia, letargia, déficit ponderal e retardo de desenvolvimento, frequentemente acompanhadas por hipotonia e cardiomiopatia. Alguns episódios de Síndrome de Morte Súbita do Lactente (SMSL) são possivelmente causados por LCHAD.
- O diagnóstico e tratamento precoces podem prevenir episódios de risco de vida. O jejum deve ser evitado e além disso, deve-se instituir uma dieta rica em carboidratos.
- Deficiência de desidrogenase de acil-CoA de cadeia média (MCAD)
- MCAD é caracterizada por episódios recorrentes de acidose metabólica, e por hipoglicemia, letargia e coma. Os sintomas tipicamente começam no lactente ou em crianças jovens.
- MCAD ocorre principalmente entre brancos descendentes do norte europeu.
- Os episódios iniciais são frequentemente desencadeados por jejum e podem levar à morte em 20 a 25 por cento dos casos.
- Aproximadamente 25% das mortes atribuídas à Síndrome de Morte Súbita do Lactente (SMSL) são, provavelmente, resultantes de MCAD.
- Evitar o jejum é imperativo. A administração de glicose IV é necessária quando houver intolerância alimentar. Ingesta elevada de ácidos graxos de cadeia média e longa deve ser evitada. Suplementos de carnitina são recomendados para estas crianças.
- Deficiência de desidrogenase de acil-CoA de cadeia curta (SCAD)
- Os sintomas iniciais de SCAD são o déficit ponderal e a hipoglicemia. Ao longo prazo, há um retardo do desenvolvimento.
- Como os outros distúrbios da oxidação dos ácidos graxos, o jejum pode precipitar uma crise e deve ser evitado.
- A dieta deve ser controlada e suplementos de carnitina devem ser administrados.
| Deficiências de enzimas do transporte e da beta-oxidação mitocondriais de ácidos graxos em humanos: Os pacientes acometidos por estas doenças podem ficar sem diagnóstico em parte porque o aparecimento de manifestações clínicas muitas vezes só acontece após jejum prolongado, e em parte devido à dificuldade do diagnóstico, pois os exames laboratoriais de rotina não ajudam o clínico a suspeitar de um defeito de beta-oxidação dos ácidos graxos. A cromatografia gasosa acoplada à espectrometria de massas (GC-MS) ou a espectrometria de massas em tandem (MS/ MS) são imprescindíveis no diagnóstico destas deficiências, especialmente em pacientes assintomáticos. |